MIT Researchers Develop ChemXploreML: A User-Friendly AI App for Chemical Property Prediction
2 Sources
2 Sources
[1]
New machine-learning application to help researchers predict chemical properties
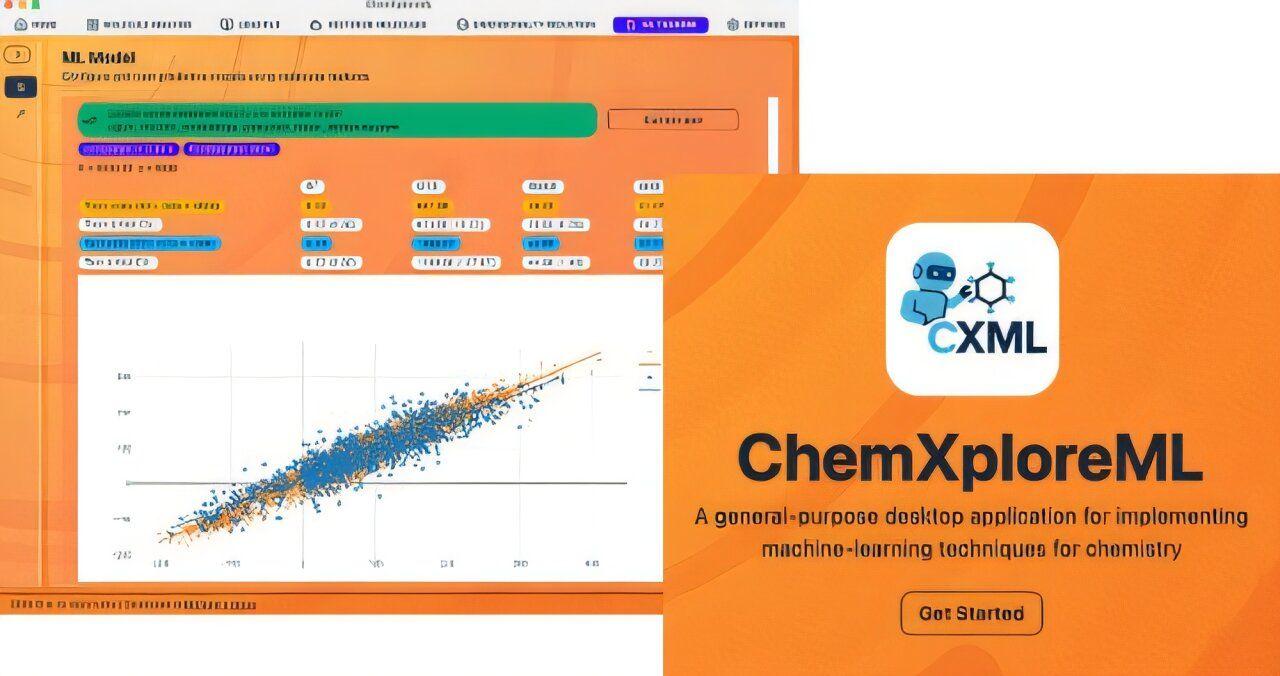
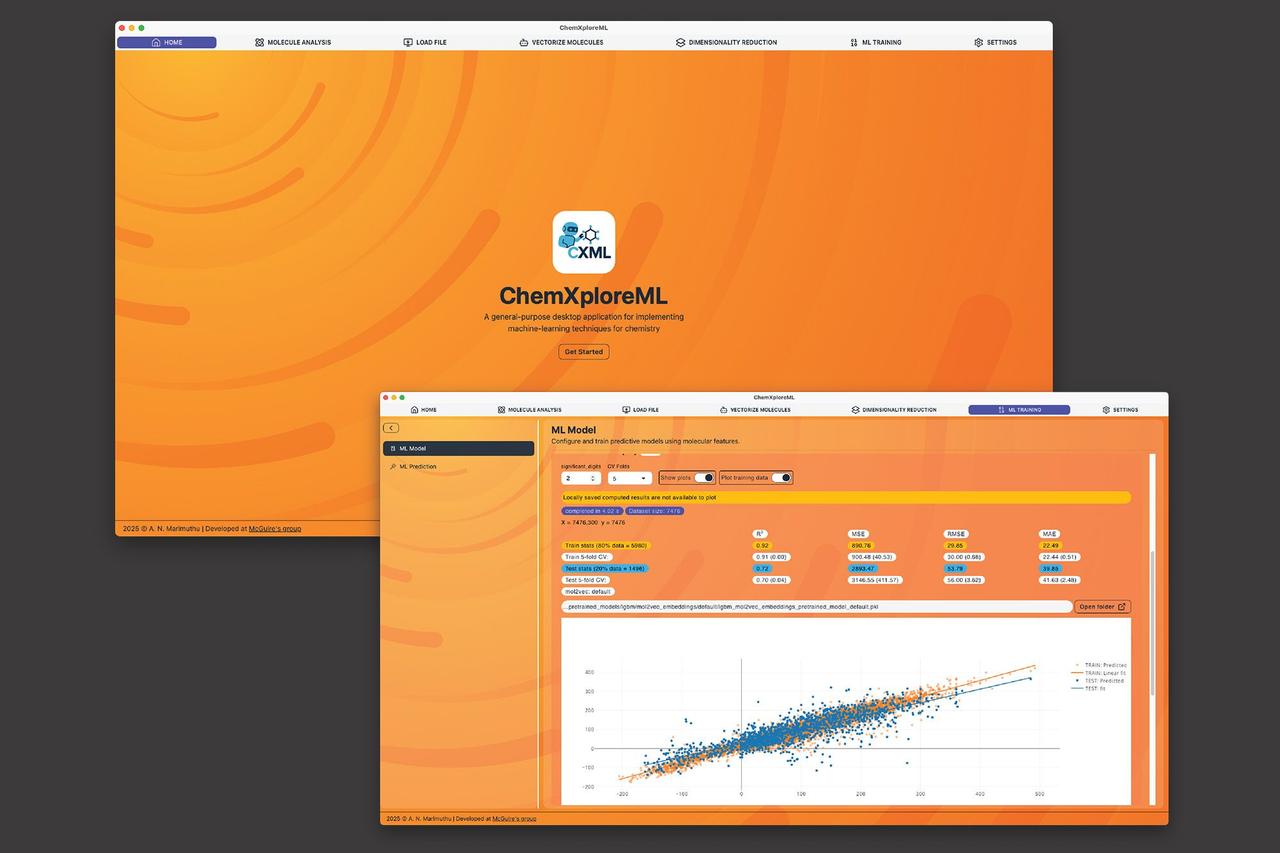
One of the shared, fundamental goals of most chemistry researchers is the need to predict a molecule's properties, such as its boiling or melting point. Once researchers can pinpoint that prediction, they're able to move forward with their work yielding discoveries that lead to medicines, materials, and more. Historically, however, the traditional methods of unveiling these predictions are associated with a significant cost -- expending time and wear and tear on equipment, in addition to funds. Enter a branch of artificial intelligence known as machine learning (ML). ML has lessened the burden of molecule property prediction to a degree, but the advanced tools that most effectively expedite the process -- by learning from existing data to make rapid predictions for new molecules -- require the user to have a significant level of programming expertise. This creates an accessibility barrier for many chemists, who may not have the significant computational proficiency required to navigate the prediction pipeline. To alleviate this challenge, researchers in the McGuire Research Group at MIT have created ChemXploreML, a user-friendly desktop app that helps chemists make these critical predictions without requiring advanced programming skills. Freely available, easy to download, and functional on mainstream platforms, this app is also built to operate entirely offline, which helps keep research data proprietary. The exciting new technology is outlined in an article published recently in the Journal of Chemical Information and Modeling. One specific hurdle in chemical machine learning is translating molecular structures into a numerical language that computers can understand. ChemXploreML automates this complex process with powerful, built-in "molecular embedders" that transform chemical structures into informative numerical vectors. Next, the software implements state-of-the-art algorithms to identify patterns and accurately predict molecular properties like boiling and melting points, all through an intuitive, interactive graphical interface. "The goal of ChemXploreML is to democratize the use of machine learning in the chemical sciences," says Aravindh Nivas Marimuthu, a postdoc in the McGuire Group and lead author of the article. "By creating an intuitive, powerful, and offline-capable desktop application, we are putting state-of-the-art predictive modeling directly into the hands of chemists, regardless of their programming background. This work not only accelerates the search for new drugs and materials by making the screening process faster and cheaper, but its flexible design also opens doors for future innovations." ChemXploreML is designed to to evolve over time, so as future techniques and algorithms are developed, they can be seamlessly integrated into the app, ensuring that researchers are always able to access and implement the most up-to-date methods. The application was tested on five key molecular properties of organic compounds -- melting point, boiling point, vapor pressure, critical temperature, and critical pressure -- and achieved high accuracy scores of up to 93 percent for the critical temperature. The researchers also demonstrated that a new, more compact method of representing molecules (VICGAE) was nearly as accurate as standard methods, such as Mol2Vec, but was up to 10 times faster. "We envision a future where any researcher can easily customize and apply machine learning to solve unique challenges, from developing sustainable materials to exploring the complex chemistry of interstellar space," says Marimuthu. Joining him on the paper is senior author and Class of 1943 Career Development Assistant Professor of Chemistry Brett McGuire.
[2]
Machine-learning application makes advanced chemical predictions easier and faster, no deep programming skills required
One of the shared, fundamental goals of most chemistry researchers is the need to predict a molecule's properties, such as its boiling or melting point. Once researchers can pinpoint that prediction, they're able to move forward with their work, yielding discoveries that lead to medicines, materials, and more. Historically, however, the traditional methods of unveiling these predictions are associated with a significant cost -- expending time and wear and tear on equipment, in addition to funds. Enter a branch of artificial intelligence known as machine learning (ML). ML has lessened the burden of molecule property prediction to a degree, but the advanced tools that most effectively expedite the process -- by learning from existing data to make rapid predictions for new molecules -- require the user to have a significant level of programming expertise. This creates an accessibility barrier for many chemists, who may not have the significant computational proficiency required to navigate the prediction pipeline. To alleviate this challenge, researchers in the McGuire Research Group at MIT have created ChemXploreML, a user-friendly desktop app that helps chemists make these critical predictions without requiring advanced programming skills. Freely available, easy to download, and functional on mainstream platforms, this app is also built to operate entirely offline, which helps keep research data proprietary. The technology is outlined in an article published recently in the Journal of Chemical Information and Modeling. One specific hurdle in chemical machine learning is translating molecular structures into a numerical language that computers can understand. ChemXploreML automates this complex process with powerful, built-in "molecular embedders" that transform chemical structures into informative numerical vectors. Next, the software implements state-of-the-art algorithms to identify patterns and accurately predict molecular properties like boiling and melting points, all through an intuitive, interactive graphical interface. "The goal of ChemXploreML is to democratize the use of machine learning in the chemical sciences," says Aravindh Nivas Marimuthu, a postdoc in the McGuire Group and lead author of the article. "By creating an intuitive, powerful, and offline-capable desktop application, we are putting state-of-the-art predictive modeling directly into the hands of chemists, regardless of their programming background. This work not only accelerates the search for new drugs and materials by making the screening process faster and cheaper, but its flexible design also opens doors for future innovations." ChemXploreML is designed to evolve over time, so as future techniques and algorithms are developed, they can be seamlessly integrated into the app, ensuring that researchers are always able to access and implement the most up-to-date methods. The application was tested on five key molecular properties of organic compounds -- melting point, boiling point, vapor pressure, critical temperature, and critical pressure -- and achieved high accuracy scores of up to 93% for the critical temperature. The researchers also demonstrated that a new, more compact method of representing molecules (VICGAE) was nearly as accurate as standard methods, such as Mol2Vec, but was up to 10 times faster. "We envision a future where any researcher can easily customize and apply machine learning to solve unique challenges, from developing sustainable materials to exploring the complex chemistry of interstellar space," says Marimuthu. Joining him on the paper was senior author and Class of 1943 Career Development Assistant Professor of Chemistry Brett McGuire.
Share
Share
Copy Link
MIT's McGuire Research Group has created ChemXploreML, a machine learning application that simplifies the prediction of chemical properties without requiring advanced programming skills, potentially revolutionizing chemical research and drug discovery.
Revolutionizing Chemical Property Prediction with AI
Researchers at MIT's McGuire Research Group have developed ChemXploreML, a groundbreaking machine learning application that simplifies the prediction of chemical properties. This innovative tool addresses a fundamental challenge in chemistry research: accurately predicting molecular properties such as boiling and melting points without the need for extensive programming expertise
1
2
.Democratizing Machine Learning in Chemistry
ChemXploreML is designed to make advanced predictive modeling accessible to chemists regardless of their computational background. The application features:
- User-friendly interface: An intuitive, interactive graphical interface that doesn't require advanced programming skills
1
. - Offline capability: Fully functional without an internet connection, ensuring data privacy and proprietary research protection
2
. - Cross-platform compatibility: Easy to download and use on mainstream platforms
1
.
Lead author Aravindh Nivas Marimuthu emphasizes that ChemXploreML aims to "democratize the use of machine learning in the chemical sciences"
1
2
.
Source: Phys.org
Advanced Features and Performance
ChemXploreML incorporates several cutting-edge features:
- Automated molecular structure translation: Built-in "molecular embedders" convert chemical structures into numerical vectors that computers can process
1
2
. - State-of-the-art algorithms: The software employs advanced algorithms to identify patterns and predict molecular properties accurately
1
. - High accuracy: Achieved up to 93% accuracy in predicting critical temperature during testing
1
2
. - Efficient molecular representation: A new method called VICGAE proved to be nearly as accurate as standard methods like Mol2Vec but up to 10 times faster
1
2
.
Related Stories
Impact on Chemical Research and Beyond
The development of ChemXploreML has significant implications for various fields:
- Accelerated drug discovery: By streamlining the screening process, the app could speed up the search for new medicines
1
2
. - Materials science advancements: Faster and cheaper property predictions could lead to breakthroughs in developing new materials
1
. - Interstellar chemistry exploration: The tool's flexibility opens up possibilities for studying complex chemical processes in space
1
2
.
Future-Proof Design
ChemXploreML is built with adaptability in mind:
- Evolving platform: The app is designed to integrate future techniques and algorithms seamlessly
1
2
. - Continuous improvement: Researchers will always have access to the most up-to-date methods
1
.
The research team, led by Marimuthu and senior author Brett McGuire, has published their findings in the Journal of Chemical Information and Modeling, detailing the potential of ChemXploreML to transform chemical research methodologies
1
2
.
Source: MIT
As machine learning continues to reshape scientific research, tools like ChemXploreML are paving the way for more efficient, accessible, and innovative approaches to solving complex chemical challenges.
References
Summarized by
Navi
Related Stories
AI in Chemistry breakthrough: Two systems accelerate molecule design and drug discovery by 10x
29 Jan 2026•Science and Research

AutoSolvateWeb: AI-Powered Chatbot Democratizes Computational Chemistry
09 Apr 2025•Science and Research

MIT Researchers Develop FlowER: A Generative AI Approach for Accurate Chemical Reaction Predictions
04 Sept 2025•Science and Research

Recent Highlights
1
Apple Plans Major Siri AI Overhaul in iOS 27 With Third-Party Chatbot Integration
Technology

2
OpenAI shuts down Sora after six months, ending Disney's $1 billion licensing partnership
Technology

3
AI chatbots validate you too much, making you less kind to others, Stanford study reveals
Science and Research

Recent Highlights
Today's Top Stories




Your Daily Dose of Curated AI News
Don’t drown in AI news. We cut through the noise - filtering, ranking and summarizing the most important AI news, breakthroughs and research daily. Spend less time searching for the latest in AI and get straight to action.