MIT Researchers Unveil Inner Workings of AI Protein Language Models
3 Sources
3 Sources
[1]
Researchers glimpse the inner workings of protein language models
Caption: Understanding what is happening inside the "black box" of large protein models could help researchers to choose better models for a particular task, helping to streamline the process of identifying new drugs or vaccine targets. Within the past few years, models that can predict the structure or function of proteins have been widely used for a variety of biological applications, such as identifying drug targets and designing new therapeutic antibodies. These models, which are based on large language models (LLMs), can make very accurate predictions of a protein's suitability for a given application. However, there's no way to determine how these models make their predictions or which protein features play the most important role in those decisions. In a new study, MIT researchers have used a novel technique to open up that "black box" and allow them to determine what features a protein language model takes into account when making predictions. Understanding what is happening inside that black box could help researchers to choose better models for a particular task, helping to streamline the process of identifying new drugs or vaccine targets. "Our work has broad implications for enhanced explainability in downstream tasks that rely on these representations," says Bonnie Berger, the Simons Professor of Mathematics, head of the Computation and Biology group in MIT's Computer Science and Artificial Intelligence Laboratory, and the senior author of the study. "Additionally, identifying features that protein language models track has the potential to reveal novel biological insights from these representations." Onkar Gujral, an MIT graduate student, is the lead author of the study, which appears this week in the Proceedings of the National Academy of Sciences. Mihir Bafna, an MIT graduate student, and Eric Alm, an MIT professor of biological engineering, are also authors of the paper. Opening the black box In 2018, Berger and former MIT graduate student Tristan Bepler PhD '20 introduced the first protein language model. Their model, like subsequent protein models that accelerated the development of AlphaFold, such as ESM2 and OmegaFold, was based on LLMs. These models, which include ChatGPT, can analyze huge amounts of text and figure out which words are most likely to appear together. Protein language models use a similar approach, but instead of analyzing words, they analyze amino acid sequences. Researchers have used these models to predict the structure and function of proteins, and for applications such as identifying proteins that might bind to particular drugs. In a 2021 study, Berger and colleagues used a protein language model to predict which sections of viral surface proteins are less likely to mutate in a way that enables viral escape. This allowed them to identify possible targets for vaccines against influenza, HIV, and SARS-CoV-2. However, in all of these studies, it has been impossible to know how the models were making their predictions. "We would get out some prediction at the end, but we had absolutely no idea what was happening in the individual components of this black box," Berger says. In the new study, the researchers wanted to dig into how protein language models make their predictions. Just like LLMs, protein language models encode information as representations that consist of a pattern of activation of different "nodes" within a neural network. These nodes are analogous to the networks of neurons that store memories and other information within the brain. The inner workings of LLMs are not easy to interpret, but within the past couple of years, researchers have begun using a type of algorithm known as a sparse autoencoder to help shed some light on how those models make their predictions. The new study from Berger's lab is the first to use this algorithm on protein language models. Sparse autoencoders work by adjusting how a protein is represented within a neural network. Typically, a given protein will be represented by a pattern of activation of a constrained number of neurons, for example, 480. A sparse autoencoder will expand that representation into a much larger number of nodes, say 20,000. When information about a protein is encoded by only 480 neurons, each node lights up for multiple features, making it very difficult to know what features each node is encoding. However, when the neural network is expanded to 20,000 nodes, this extra space along with a sparsity constraint gives the information room to "spread out." Now, a feature of the protein that was previously encoded by multiple nodes can occupy a single node. "In a sparse representation, the neurons lighting up are doing so in a more meaningful manner," Gujral says. "Before the sparse representations are created, the networks pack information so tightly together that it's hard to interpret the neurons." Interpretable models Once the researchers obtained sparse representations of many proteins, they used an AI assistant called Claude (related to the popular Anthropic chatbot of the same name), to analyze the representations. In this case, they asked Claude to compare the sparse representations with the known features of each protein, such as molecular function, protein family, or location within a cell. By analyzing thousands of representations, Claude can determine which nodes correspond to specific protein features, then describe them in plain English. For example, the algorithm might say, "This neuron appears to be detecting proteins involved in transmembrane transport of ions or amino acids, particularly those located in the plasma membrane." This process makes the nodes far more "interpretable," meaning the researchers can tell what each node is encoding. They found that the features most likely to be encoded by these nodes were protein family and certain functions, including several different metabolic and biosynthetic processes. "When you train a sparse autoencoder, you aren't training it to be interpretable, but it turns out that by incentivizing the representation to be really sparse, that ends up resulting in interpretability," Gujral says. Understanding what features a particular protein model is encoding could help researchers choose the right model for a particular task, or tweak the type of input they give the model, to generate the best results. Additionally, analyzing the features that a model encodes could one day help biologists to learn more about the proteins that they are studying. "At some point when the models get a lot more powerful, you could learn more biology than you already know, from opening up the models," Gujral says. The research was funded by the National Institutes of Health.
[2]
Researchers glimpse the inner workings of protein language models
Within the past few years, models that can predict the structure or function of proteins have been widely used for a variety of biological applications, such as identifying drug targets and designing new therapeutic antibodies. These models, which are based on large language models (LLMs), can make very accurate predictions of a protein's suitability for a given application. However, there's no way to determine how these models make their predictions or which protein features play the most important role in those decisions. In a new study, MIT researchers have used a novel technique to open up that "black box" and allow them to determine what features a protein language model takes into account when making predictions. Understanding what is happening inside that black box could help researchers to choose better models for a particular task, helping to streamline the process of identifying new drugs or vaccine targets. "Our work has broad implications for enhanced explainability in downstream tasks that rely on these representations," says Bonnie Berger, the Simons Professor of Mathematics, head of the Computation and Biology group in MIT's Computer Science and Artificial Intelligence Laboratory, and the senior author of the study. "Additionally, identifying features that protein language models track has the potential to reveal novel biological insights from these representations." Onkar Gujral, an MIT graduate student, is the lead author of the study, which appears this week in the Proceedings of the National Academy of Sciences. Mihir Bafna, an MIT graduate student, and Eric Alm, an MIT professor of biological engineering, are also authors of the paper. Opening the black box In 2018, Berger and former MIT graduate student Tristan Bepler, Ph.D. introduced the first protein language model. Their model, like subsequent protein models that accelerated the development of AlphaFold, such as ESM2 and OmegaFold, was based on LLMs. These models, which include ChatGPT, can analyze huge amounts of text and figure out which words are most likely to appear together. Protein language models use a similar approach, but instead of analyzing words, they analyze amino acid sequences. Researchers have used these models to predict the structure and function of proteins, and for applications such as identifying proteins that might bind to particular drugs. In a 2021 study, Berger and colleagues used a protein language model to predict which sections of viral surface proteins are less likely to mutate in a way that enables viral escape. This allowed them to identify possible targets for vaccines against influenza, HIV, and SARS-CoV-2. However, in all of these studies, it has been impossible to know how the models were making their predictions. "We would get out some prediction at the end, but we had absolutely no idea what was happening in the individual components of this black box," Berger says. In the new study, the researchers wanted to dig into how protein language models make their predictions. Just like LLMs, protein language models encode information as representations that consist of a pattern of activation of different "nodes" within a neural network. These nodes are analogous to the networks of neurons that store memories and other information within the brain. The inner workings of LLMs are not easy to interpret, but within the past couple of years, researchers have begun using a type of algorithm known as a sparse autoencoder to help shed some light on how those models make their predictions. The new study from Berger's lab is the first to use this algorithm on protein language models. Sparse autoencoders work by adjusting how a protein is represented within a neural network. Typically, a given protein will be represented by a pattern of activation of a constrained number of neurons, for example, 480. A sparse autoencoder will expand that representation into a much larger number of nodes, say 20,000. When information about a protein is encoded by only 480 neurons, each node lights up for multiple features, making it very difficult to know what features each node is encoding. However, when the neural network is expanded to 20,000 nodes, this extra space along with a sparsity constraint gives the information room to "spread out." Now, a feature of the protein that was previously encoded by multiple nodes can occupy a single node. "In a sparse representation, the neurons lighting up are doing so in a more meaningful manner," Gujral says. "Before the sparse representations are created, the networks pack information so tightly together that it's hard to interpret the neurons." Interpretable models Once the researchers obtained sparse representations of many proteins, they used an AI assistant called Claude (related to the popular Anthropic chatbot of the same name), to analyze the representations. In this case, they asked Claude to compare the sparse representations with the known features of each protein, such as molecular function, protein family, or location within a cell. By analyzing thousands of representations, Claude can determine which nodes correspond to specific protein features, then describe them in plain English. For example, the algorithm might say, "This neuron appears to be detecting proteins involved in transmembrane transport of ions or amino acids, particularly those located in the plasma membrane." This process makes the nodes far more "interpretable," meaning the researchers can tell what each node is encoding. They found that the features most likely to be encoded by these nodes were protein family and certain functions, including several different metabolic and biosynthetic processes. "When you train a sparse autoencoder, you aren't training it to be interpretable, but it turns out that by incentivizing the representation to be really sparse, that ends up resulting in interpretability," Gujral says. Understanding what features a particular protein model is encoding could help researchers choose the right model for a particular task, or tweak the type of input they give the model, to generate the best results. Additionally, analyzing the features that a model encodes could one day help biologists to learn more about the proteins that they are studying. "At some point when the models get a lot more powerful, you could learn more biology than you already know, from opening up the models," Gujral says.
[3]
MIT technique reveals how AI models predict protein functions
Massachusetts Institute of TechnologyAug 19 2025 Within the past few years, models that can predict the structure or function of proteins have been widely used for a variety of biological applications, such as identifying drug targets and designing new therapeutic antibodies. These models, which are based on large language models (LLMs), can make very accurate predictions of a protein's suitability for a given application. However, there's no way to determine how these models make their predictions or which protein features play the most important role in those decisions. In a new study, MIT researchers have used a novel technique to open up that "black box" and allow them to determine what features a protein language model takes into account when making predictions. Understanding what is happening inside that black box could help researchers to choose better models for a particular task, helping to streamline the process of identifying new drugs or vaccine targets. Our work has broad implications for enhanced explainability in downstream tasks that rely on these representations. Additionally, identifying features that protein language models track has the potential to reveal novel biological insights from these representations." Bonnie Berger, Study Senior Author and Simons Professor of Mathematics, Massachusetts Institute of Technology Berger is also the head of the Computation and Biology group in MIT's Computer Science and Artificial Intelligence Laboratory. Onkar Gujral, an MIT graduate student, is the lead author of the study, which appears this week in the Proceedings of the National Academy of Sciences. Mihir Bafna, an MIT graduate student, and Eric Alm, an MIT professor of biological engineering, are also authors of the paper. Opening the black box In 2018, Berger and former MIT graduate student Tristan Bepler PhD '20 introduced the first protein language model. Their model, like subsequent protein models that accelerated the development of AlphaFold, such as ESM2 and OmegaFold, was based on LLMs. These models, which include ChatGPT, can analyze huge amounts of text and figure out which words are most likely to appear together. Protein language models use a similar approach, but instead of analyzing words, they analyze amino acid sequences. Researchers have used these models to predict the structure and function of proteins, and for applications such as identifying proteins that might bind to particular drugs. In a 2021 study, Berger and colleagues used a protein language model to predict which sections of viral surface proteins are less likely to mutate in a way that enables viral escape. This allowed them to identify possible targets for vaccines against influenza, HIV, and SARS-CoV-2. However, in all of these studies, it has been impossible to know how the models were making their predictions. "We would get out some prediction at the end, but we had absolutely no idea what was happening in the individual components of this black box," Berger stated. In the new study, the researchers wanted to dig into how protein language models make their predictions. Just like LLMs, protein language models encode information as representations that consist of a pattern of activation of different "nodes" within a neural network. These nodes are analogous to the networks of neurons that store memories and other information within the brain. The inner workings of LLMs are not easy to interpret, but within the past couple of years, researchers have begun using a type of algorithm known as a sparse autoencoder to help shed some light on how those models make their predictions. The new study from Berger's lab is the first to use this algorithm on protein language models. Sparse autoencoders work by adjusting how a protein is represented within a neural network. Typically, a given protein will be represented by a pattern of activation of a constrained number of neurons, for example, 480. A sparse autoencoder will expand that representation into a much larger number of nodes, say 20,000. When information about a protein is encoded by only 480 neurons, each node lights up for multiple features, making it very difficult to know what features each node is encoding. However, when the neural network is expanded to 20,000 nodes, this extra space along with a sparsity constraint gives the information room to "spread out." Now, a feature of the protein that was previously encoded by multiple nodes can occupy a single node. "In a sparse representation, the neurons lighting up are doing so in a more meaningful manner," Gujral says. "Before the sparse representations are created, the networks pack information so tightly together that it's hard to interpret the neurons." Interpretable models Once the researchers obtained sparse representations of many proteins, they used an AI assistant called Claude (related to the popular Anthropic chatbot of the same name), to analyze the representations. In this case, they asked Claude to compare the sparse representations with the known features of each protein, such as molecular function, protein family, or location within a cell. By analyzing thousands of representations, Claude can determine which nodes correspond to specific protein features, then describe them in plain English. For example, the algorithm might say, "This neuron appears to be detecting proteins involved in transmembrane transport of ions or amino acids, particularly those located in the plasma membrane." This process makes the nodes far more "interpretable," meaning the researchers can tell what each node is encoding. They found that the features most likely to be encoded by these nodes were protein family and certain functions, including several different metabolic and biosynthetic processes. "When you train a sparse autoencoder, you aren't training it to be interpretable, but it turns out that by incentivizing the representation to be really sparse, that ends up resulting in interpretability," Gujral says. Understanding what features a particular protein model is encoding could help researchers choose the right model for a particular task, or tweak the type of input they give the model, to generate the best results. Additionally, analyzing the features that a model encodes could one day help biologists to learn more about the proteins that they are studying. "At some point when the models get a lot more powerful, you could learn more biology than you already know, from opening up the models," Gujral says. Massachusetts Institute of Technology
Share
Share
Copy Link
MIT scientists have developed a novel technique to understand how AI protein language models make predictions, potentially streamlining drug discovery and vaccine development processes.
Unveiling the Black Box of Protein Language Models
Researchers at the Massachusetts Institute of Technology (MIT) have made a significant breakthrough in understanding the inner workings of protein language models, a type of artificial intelligence used in various biological applications. The study, published in the Proceedings of the National Academy of Sciences, introduces a novel technique to decipher how these models make predictions about protein structure and function
1
.
Source: News-Medical
The Challenge of Interpretability
Protein language models, based on large language models (LLMs), have been widely used in recent years for tasks such as identifying drug targets and designing therapeutic antibodies. While these models can make accurate predictions, they have long been considered "black boxes," with researchers unable to determine how they arrive at their conclusions
2
.A Novel Approach: Sparse Autoencoders
The MIT team, led by Bonnie Berger, the Simons Professor of Mathematics and head of the Computation and Biology group at MIT's Computer Science and Artificial Intelligence Laboratory, employed a technique called sparse autoencoders to open up this black box
3
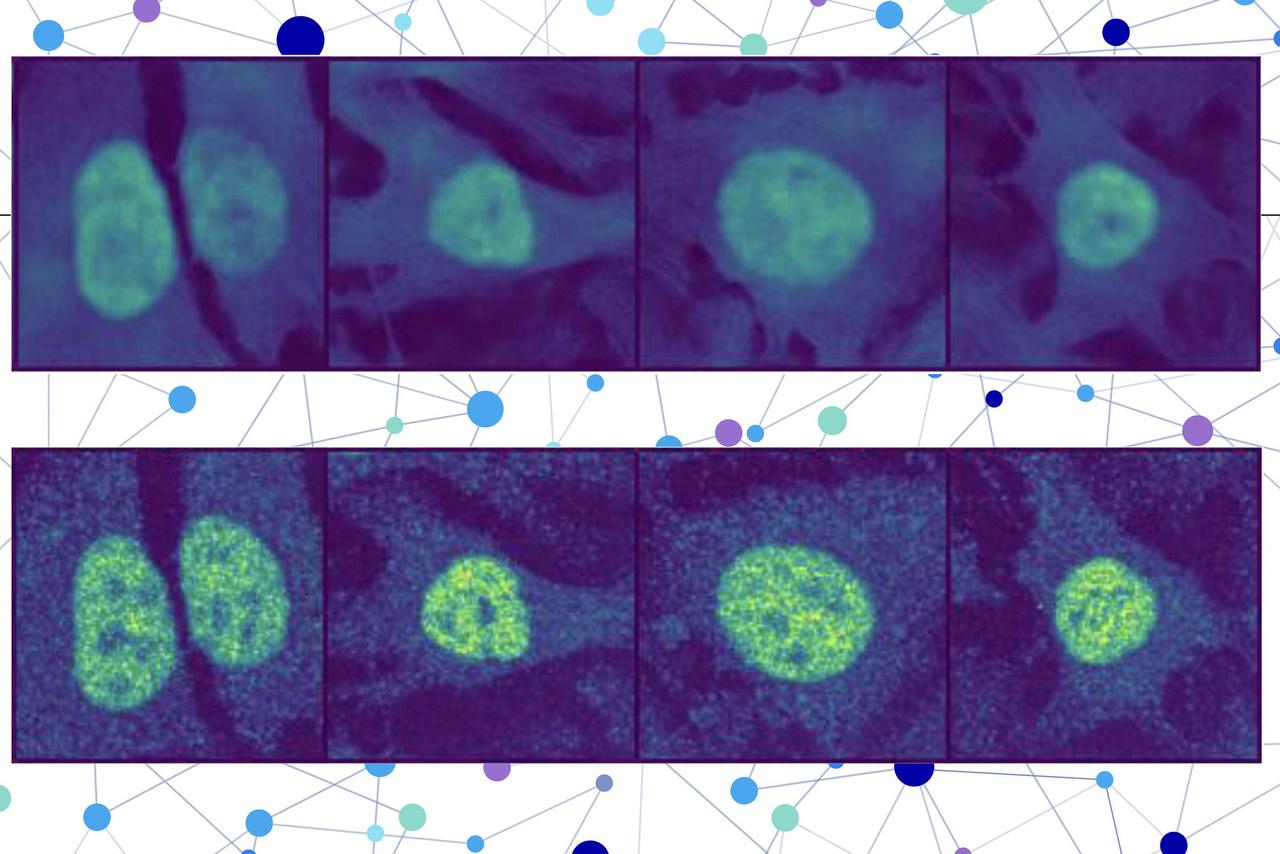
.Sparse autoencoders work by expanding the neural network representation of a protein from a constrained number of neurons (e.g., 480) to a much larger number (e.g., 20,000). This expansion allows the information to "spread out," making it easier to interpret which features each node is encoding
1
.
Source: MIT
Interpreting the Results with AI Assistance
To analyze the expanded representations, the researchers utilized an AI assistant called Claude. By comparing the sparse representations with known protein features, Claude could determine which nodes corresponded to specific protein characteristics and describe them in plain English
2
.Related Stories
Key Findings and Implications
The study revealed that the features most likely to be encoded by these nodes were protein family and certain functions, including various metabolic and biosynthetic processes. This insight into how protein language models make predictions could have significant implications for biological research and drug development
3
.Historical Context and Future Potential
The first protein language model was introduced in 2018 by Berger and former MIT graduate student Tristan Bepler. Since then, these models have been used for various applications, including predicting viral protein mutations to identify vaccine targets for influenza, HIV, and SARS-CoV-2
1
.By making these models more interpretable, researchers can potentially choose better models for specific tasks, streamlining the process of identifying new drugs or vaccine targets. As Berger notes, "Our work has broad implications for enhanced explainability in downstream tasks that rely on these representations"
2
.References
Summarized by
Navi
[3]
Related Stories
AI Revolutionizes Protein Sequencing: InstaNovo Models Enhance Accuracy and Discovery
31 Mar 2025•Science and Research

OpenFold3 Emerges as Open-Source Alternative to AlphaFold3, Democratizing Protein Structure Prediction
29 Oct 2025•Science and Research

AlphaFold Upgrade: AI Now Predicts Large Protein Structures and Integrates Experimental Data
05 Nov 2024•Science and Research

Recent Highlights
1
OpenAI shuts down Sora video app after six months, ending Disney's $1 billion investment deal
Technology

2
AI-Generated Val Kilmer to Posthumously Appear in As Deep as the Grave After His Death
Entertainment and Society

3
Supermicro Co-Founder Indicted in $2.5 Billion Nvidia AI Chip Smuggling Scheme to China
Policy and Regulation

Recent Highlights
Today's Top Stories




Your Daily Dose of Curated AI News
Don’t drown in AI news. We cut through the noise - filtering, ranking and summarizing the most important AI news, breakthroughs and research daily. Spend less time searching for the latest in AI and get straight to action.