Harvard's AI model predicts immunotherapy outcomes across cancer types with improved accuracy
2 Sources
[1]
Generalizable AI predicts immunotherapy outcomes across cancers and treatments - Nature Medicine
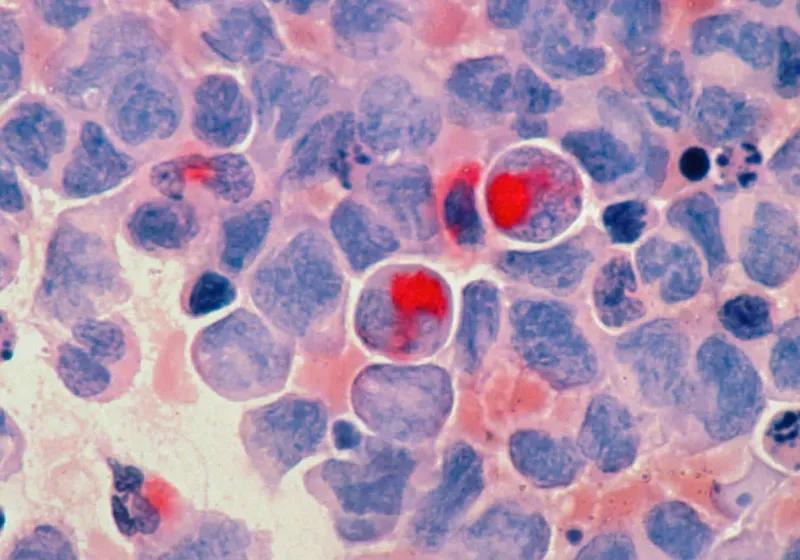
COMPASS links tumor transcriptomes to interpretable immune representations and supports biomarker discovery, mechanistic hypothesis generation and patient stratification in immunotherapy trials. Its performance across cancer types and checkpoint inhibitor therapies supports the use of mechanistically interpretable immune modeling in translational research and clinical development. This study is a computational analysis using deidentified datasets. No new patient data were collected, and no ethical approval from an institutional review board or ethics committee was required. This section describes the following: (1) dataset curation and preprocessing, (2) the COMPASS model, (3) self-supervised pretraining of COMPASS, (4) supervised fine-tuning for response prediction, (5) benchmarking COMPASS models against established methods, (6) MSFT for drug-specific and disease-specific models, (7) SHapley Additive exPlanations (SHAP) analysis of important features, (8) overall survival analysis, (9) TIME concept analysis in the IMvigor210 cohort and (10) personalized response maps generation. Dataset curation and processing TCGA datasets Pretraining datasets were acquired from TCGA via the Genomic Data Commons (GDC) portal (version 37; GDC Portal), using TCGAbiolinks for data retrieval. To ensure cross-cohort compatibility with downstream ICI analyses, all RNA-seq data are uniformly processed through our standardized pipeline. Read alignment was performed against the GRCh38/hg38 reference genome using STAR (version 2.7.5c), with gene features annotated according to GENCODE version 36. Raw counts are normalized by gene effective length and converted to transcripts per million (TPM): where is the number of genes and This normalization facilitates cross-sample comparability. Initial data included 60,660 genes across 11,274 samples. After excluding normal tissue samples, 10,534 samples remained. Further exclusions were applied for previous treatment samples and non-formalin-fixed paraffin-embedded (FFPE) samples, resulting in 10,305 samples. Finally, aggregation to the patient level using the 'bcr patient barcode' key yielded 10,184 unique patient tumor samples. Protein-coding genes are selected (15,672 genes) based on overlap with the gene expression data from the clinical cohorts. ICI clinical cohorts We curate 16 cohorts (Fig. 2a) spanning seven cancer types, categorized into three groups: large cohorts (>100 patients), medium-sized cohorts (30-100 patients) and small cohorts (<30 patients). Publicly available RNA-seq data underwent uniform processing through the same standardized pipeline that was used to process the TCGA data (see previous section), converting raw sequencing data (FASTQ) to counts and TPM values. For cohorts with available raw data, FASTQ files were reprocessed; otherwise, TPM values were derived from the counts using TCGA-aligned gene lengths. To ensure cross-cohort consistency, all data were mapped to the same reference genome, correcting for potential differences in original genomic builds. To ensure reproducibility, researchers may download raw data using accession IDs in Extended Data Table 1 and reprocess them via our publicly available code (https://github.com/mims-harvard/COMPASS-web/tree/main/mRNA_pipeline), which includes parameters for alignment, quantification and TPM conversion. All steps rely on the GRCh38 reference genome and GENCODE version 36 annotations to maintain cross-cohort consistency. Only pretreatment samples are included. Responders are defined as patients achieving partial response or complete response, and non-responders include those with stable disease or progressive disease, per Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST v1.1) best overall response (BOR) criteria as reported in source studies, unless otherwise noted. See Extended Data Table 1 for cohort overview. Large immunotherapy cohorts The IMvigor210 cohort (n = 298) includes patients with atezolizumab-treated bladder cancer (BLCA) (68 responders, 230 non-responders), with data sourced from the IMvigor210CoreBiologies (version 1.0.1) R package and Cancer Research Institute (CRI) iAtlas. The IMmotion150 cohort (n = 165) comprises patients with atezolizumab-treated clear-cell renal cell carcinoma (KIRC) (48 responders, 117 non-responders). For the cohort from Liu et al. (n = 107), posttreatment samples are excluded, retaining 41 patients with melanoma (nivolumab/pembrolizumab) classified as responders and 66 classified as non-responders. The Ravi-1 cohort (n = 102) is a subcohort of the SU2C-MARK non-small cell lung cancer (NSCLC) study, focusing on patients with LUAD receiving PD-(L)1 ± CTLA-4 inhibitors. Medium-sized immunotherapy cohorts The Rose et al. cohort (n = 89) includes patients with BLCA treated with PD-(L)1 inhibitors (16 responders, 73 non-responders). The Gide et al. cohort (n = 73) comprises patients with melanoma receiving anti-PD-1 ± anti-CTLA-4 (40 responders, 33 non-responders). Additional cohorts include Riaz et al. (n = 51) with nivolumab-treated patients with melanoma (10 responders, 41 non-responders); Kim et al. (n = 45) with pembrolizumab-treated patients with stomach adenocarcinoma (STAD) (12 responders, 33 non-responders); Van Allen et al. (n = 39) with ipilimumab-treated patients with melanoma (26 responders (complete response/partial response or stable disease with overall survival >1 year) and 13 non-responders (progressive disease or stable disease with overall survival <1 year)); and Freeman et al. (n = 34) with patients with melanoma from the MGH cohort treated with nivolumab, pembrolizumab, ipilimumab or combination therapies (12 responders, 22 non-responders). Small immunotherapy cohorts The Hugo et al. cohort (n = 26) involves pembrolizumab-treated patients with melanoma (14 responders and 12 non-responders by immune-related RECIST (irRECIST)). The Ravi-2 cohort (n = 25) represents a SU2C-MARK NSCLC substudy of patients with lung squamous cell carcinoma (LUSC) treated with PD-1 or PD-L1 inhibitors (eight responders, 17 non-responders). For the Zhao et al. cohort (n = 25), patients with glioblastoma (GBM) receiving nivolumab or pembrolizumab are classified as responders based on either (1) posttreatment histopathology showing inflammatory response with minimal/no residual tumor cells or (2) radiographic evidence of stable/shrinking tumor volume over 6 months. The Snyder et al. cohort (n = 21) includes atezolizumab-treated patients with BLCA (seven responders, 14 non-responders). For patients with KIRC: the Choueiri et al. cohort (n = 16) contains nivolumab-treated cases (three responders, 13 non-responders), whereas the Miao et al. cohort (n = 17) includes patients receiving PD-(L)1 ± CTLA-4 inhibitors (five responders, 12 non-responders), both sourced from CRI iAtlas. No new datasets were generated in this study. COMPASS model The COMPASS model comprises three key components. The first component, a transformer-based gene language model (GLM), serves as the 'encoder' to generate contextualized representations of individual genes. Next, a hierarchical 'projector' transforms these gene-level embeddings into high-level biological concepts, including immune cell types and pathways. The final component, a 'classifier', performs immunotherapy response prediction from the concept representations, employing either a multilayer perceptron (MLP) or a non-parametric, similarity-based method for zero-shot prediction. GLM encoder The GLM adapts natural language modeling techniques to transcriptomic data, where each gene is treated as a token. Unlike natural language where tokens follow a clear sequential structure, gene expression profiles are inherently unordered and represented in tabular format. This difference renders positional encodings, such as fixed sinusoidal encodings used in natural language processing, suboptimal for capturing gene-gene relationships. Drawing inspiration from FT-Transformer, which is designed for tabular data, we introduce a learnable gene-specific positional bias that enables the model to capture contextual interactions between genes in a biologically informed, data-driven manner. Gene abundance embedding Let denote the input gene expression matrix, where is the batch size and is the number of genes. Each gene is embedded into a d-dimensional latent space using a learnable embedding matrix , initialized from a uniform distribution: To generate expression-aware embeddings, we scale each geneʼs embedding vector by its corresponding expression value. Specifically, the embedding for the l-th gene in the b-th sample is given by: for and , resulting in the embedding tensor . This design enables the model to capture both gene identity (via W) and sample-specific abundance (via ) in the representation. Learnable positional encoding To inject gene-specific inductive biases into the model, we introduce a learnable positional encoding matrix , initialized in the same manner: Each gene receives a unique, trainable positional vector , which acts as a contextual bias. The final input embedding for each gene in each sample is computed by element-wise addition of the positional encoding: resulting in . Unlike fixed encodings, this learnable scheme allows the model to adaptively encode gene-level functional relevance during training, thereby serving as a gene-aware bias that enhances the transformerʼs capacity to model context-specific gene interactions. Cancer type token embedding To account for pan-cancer heterogeneity, COMPASS integrates a cancer type token that interacts with gene tokens through attention mechanisms and is separately projected as a concept in the modelʼs hierarchy. To generate the cancer type token embedding, the 33 cancer types are first encoded as integers (0-32). This integer encoding serves as an index for looking up a learnable embedding matrix: Given a batch of cancer type labels , we perform a lookup to obtain their embeddings: These embeddings are reshaped as and prepended to the gene embeddings along the sequence axis. This cancer type token interacts with gene tokens via self-attention and is later projected into a dedicated concept node in the concept hierarchy. As a robustness check, we ablated the cancer type concept during fine-tuning and reevaluated leave-one-indication-out and leave-one-target-out generalization; performance decreased moderately but remained substantial (Supplementary Fig. 10). Transformer encoder for contextual learning The full input to the transformer encoder is constructed by concatenating the cancer type token and gene embeddings: This tensor passes through a multilayer transformer encoder composed of stacked self-attention and feedforward layers: Within each layer, the self-attention mechanism enables each token to dynamically attend to all others, including gene-gene-type and gene-cancer-type interactions. The attention weights are computed via: Given the large number of genes typically used in the model, full attention becomes computationally expensive. We use the Performer architecture, which replaces standard attention with a linear approximation while preserving expressiveness. The architecture supports flash attention as an optional alternative to further reduce memory overhead and runtime. The output tensor encodes the contextualized representations of both the gene expression profile and cancer type context. Rather than directly using these latent features for prediction, we project them into an explicit, biology-grounded concept bottleneck via a hierarchical projector. The resulting interpretable concept representation serves as the exclusive input to the downstream response classifier. Concept-based hierarchical projector COMPASS follows the concept bottleneck paradigm, in which inputs pass through a layer of human-interpretable concepts rather than mapping directly from latent features to predictions. In concept bottleneck models, inputs map to a concept vector, and predictions are computed from these concepts, which support interpretability and concept-level intervention. COMPASS introduces this architecture for cancer transcriptomics by leveraging immunological gene sets and a hierarchical gene → gene set → concept mapping to represent each patientʼs TIME. To transform gene embeddings into interpretable features, COMPASS uses a hierarchical projector with two outputs: (1) gene set scores, , where each score corresponds to a curated gene set, such as a pathway or immune cell signature; and (2) concept scores, , which aggregate gene sets into functional modules, such as immune activation or immune suppression. Granular concept (gene set) aggregation Given a curated gene set with genes, we extract their contextualized embeddings from the GLM output : To aggregate the gene vectors into a unified representation, we introduce a learnable attention vector , initialized from a normal distribution and normalized via a softmax transformation: The attention-weighted gene set embedding is then computed as: This aggregated vector is passed through a linear layer to produce the scalar score for gene set : The full set of gene sets yields a tensor , where denotes the total number of gene sets in the model. This internal latent representation captures modular biological information across curated pathways and cell types. High-level concept aggregation Each high-level concept consists of a subset of gene sets , where is the number of gene sets associated with concept . The corresponding gene set scores are aggregated using a second-level attention mechanism. A learnable attention vector is normalized via a softmax transformation: The high-level concept score is then computed as: This attention-based aggregation allows each high-level concept to dynamically weight its constituent gene sets, enabling flexible and interpretable summarization of complex biological programs. Final concept representation COMPASS defines high-level concepts derived from immune-related pathways, cell types and functional groups. One additional concept represents the cancer type. The final concept representation for a given patient is: Here, the cancer type score is computed by projecting the cancer token embedding through a linear layer: Together, provides a biologically grounded and interpretable embedding of each patientʼs tumor microenvironment, suitable for downstream prediction and mechanistic analysis. Prediction module Prediction module in the COMPASS model performs the conversion of high-level concept representations into probabilistic predictions of immunotherapy response. To accommodate both standard supervised learning and generalization to new cohorts or cancer types, COMPASS supports two distinct classifier types: a parametric MLP and a non-parametric cosine similarity-based prototypical network. The latter is referred to as the NFT (no fine-tuning) classifier, as it operates without gradient-based optimization during inference. These classifier heads provide complementary strengths and can be selected based on the availability of training labels and the desired generalization behavior. Parametric classifier using an MLP The MLP classifier transforms high-level concept vectors into binary predictions via a trainable feedforward network. Given an input matrix , where is the batch size and 44 is the number of concepts (43 biological concepts plus one cancer type), the inputs are first standardized: The normalized vectors are then passed through fully connected layers, producing output logits : To calibrate output confidence, the logits are scaled by a learnable temperature parameter , defined as : These scaled logits are transformed into probabilities using the softmax function: The learnable temperature allows the model to adjust the sharpness of its predictions and improves its ability to distinguish ambiguous cases, such as borderline responders. Non-parametric classifier using prototypes The NFT classifier adopts a prototypical network architecture that performs inference through similarity comparisons with labeled support examples, eliminating the need for model fine-tuning. This non-parametric approach is advantageous when limited training data prevent effective model adaptation. As illustrated in Supplementary Fig. 1a, the classifier begins with a support set of labeled patient embeddings, each represented by a high-level concept vector and a binary response label. The support examples are grouped by class c ∈ {responder, non-responder}, and a class prototype is computed by averaging the normalized vectors in each group: where is the concept vector of the i-th support sample in class , and is the number of support examples in that class. The resulting prototypes are unit normalized to enable cosine-based comparison. Given a query patient with concept vector , the classifier computes the cosine similarity between the query and each class prototype: The similarity scores are scaled by a fixed temperature (typically 0.1) and then passed through a softmax layer: Pretraining COMPASS on TCGA dataset The COMPASS model was pretrained on bulk RNA-seq data from 10,184 patients across 33 TCGA cancer types using a self-supervised triplet contrastive learning approach. This framework learns to map tumor transcriptomes (TPM values) into a 44-dimensional concept embedding space that captures TIME features. During training, each triplet consists of an anchor sample (a patientʼs transcriptome), a positive sample (an augmented version of the same transcriptome) and a negative sample (a transcriptome from a different patient within the same cancer type). The model optimizes the embedding space to minimize cosine distance between anchor-positive pairs while maximizing separation from negative samples. To address imbalance in TCGA cohort sizes across cancer types, we implemented balanced sampling with replacement, upweighting underrepresented cancer types during training. This ensures that all cancer types contribute proportionally to the learned representations. Data augmentation For contrastive learning, each anchor transcriptome undergoes stochastic transformation via one of two augmentation methods. Random masking independently zeros each gene's expression value x with probability according to: Alternatively, Gaussian jitter adds normally distributed noise to each value: The augmentation method is selected randomly for each transformation event. We use random masking and Gaussian jitter, two widely used augmentations for contrastive learning on high-dimensional expression data. These augmentations promote robustness to technical variability, such as measurement noise. By analogy, computer vision uses domain-relevant augmentations such as rotation, cropping and brightness perturbation. Future work could develop biologically grounded augmentation strategies for RNA-seq. Sensitivity analyses showed that increasing masking probability or jitter magnitude reduced frozen-representation (NFT) performance but improved PFT performance, consistent with stronger perturbations acting as a form of representation regularization. Restricting negative sampling to local transcriptomic neighborhoods (smaller K) improved both NFT and PFT performance, supporting the importance of biologically structured hard-negative sampling (Supplementary Figs. 36 and 37 and Supplementary Methods 5). Self-supervised training The model is trained using a margin-based triplet loss function: where , and represent anchor, positive and negative COMPASS model embeddings respectively; cos denotes cosine similarity; and a default margin of 1 is used. Training was conducted on NVIDIA Tesla A100 80GB GPUs with batch size 128 and learning rate 1 × 10 using the Adam optimizer. We reserved 1% of samples as a validation set for early stopping, with training halted if validation loss failed to improve for 10 consecutive epochs. For robustness, we performed three independent training runs with random seeds 24, 42 and 64, selecting the checkpoint with lowest validation loss. Fine-tuning COMPASS for response prediction After pretraining on TCGA, the COMPASS model is fine-tuned on ICI-treated cohorts to predict clinical response. To accommodate varying dataset sizes and quality across cohorts, we implemented four complementary fine-tuning strategies with differing levels of parameter adaptation: non-parametric zero-shot inference (COMPASS-NFT mode), linear probing (COMPASS-LFT mode), partial fine-tuning (COMPASS-PFT mode) and full model fine-tuning (COMPASS-FFT mode). These approaches provide a spectrum from maximal parameter efficiency (COMPASS-NFT) to full model adaptability (COMPASS-FFT). All parametric modes (COMPASS-LFT, COMPASS-PFT and COMPASS-FFT) process the 44-dimensional concept vector through a single-layer dense classifier with 16 hidden units, generating logit outputs for binary response prediction. The trainable parameters vary substantially across modes: COMPASS-FFT updates all model parameters (approximately 1,018,784 total), including the GLM encoder and projector; COMPASS-PFT adapts only the classifier and projection layers (2,144 parameters); COMPASS-LFT modifies only the classifier head (182 parameters); and COMPASS-NFT maintains frozen pretrained weights with no trainable parameters, instead using prototypical inference based on cosine similarity in concept space. For parametric modes, models are optimized using cross-entropy loss with learning rates between 10 and 10 (slightly higher than the pretraining learning rate to promote faster adaptation to new domains), batch sizes of 8-16 (scaled to cohort size and GPU memory) and weight decay (10 and 10) tuned per mode and dataset. Internal cross-validation determined optimal training epochs, with FFT typically converging faster but being more prone to overfitting compared to the more stable LFT and PFT approaches in small datasets. All experiments ran on NVIDIA Tesla V100 GPUs, with final model selection based on cross-validated validation performance. The COMPASS-NFT mode represents a distinct non-parametric approach where labeled samples from the training cohort serve as a support set to compute responder/non-responder prototypes in the frozen 44-dimensional concept space. New samples are classified by cosine similarity to these prototypes. This enables generalization to new domains without any additional gradient updates, making COMPASS-NFT ideal for low-data or zero-shot transfer scenarios. Benchmarking immunotherapy response prediction models Overview of existing methods We evaluated COMPASS against 22 ICI response prediction methods (Supplementary Table 2). These methods encompass three main categories: (1) target gene markers (PD-1, PD-L1, CTLA-4 and their combination (GeneBio)); (2) immune cell and functional signatures including Cytotoxic Immune Signature (CIS), T-effector-IFNγ Signature (Teff), Neoadjuvant Response Signature (NRS), IFNγ Signature Score (IFNG), Cytotoxic T Lymphocyte Markers (CTL), Tumor-Associated Macrophages (TAM), T Cell Exhaustion (Texh), Chemokine Signature Score (CKS), Cancer-Associated Fibroblast Signature Score (CAF), Roh Immune Score (IS), Immune Cytolytic Activity Score (ICA), CD8 Signature Score (CD8), MHC I Association Immune Score (MIAS) and T Cell-Inflamed Gene Expression Profile Score (GEP); and (3) comprehensive integrative methods including Tumor Immune Dysfunction and Exclusion Score (TIDE), Immuno-Predictive Score (IMPRES), Paired Gene Markers (PGM) and Network-Based ICI Treatment Biomarkers (NetBio). For benchmarking, we implemented logistic regression models using the marker gene or predictive score from each method (detailed in Supplementary Table 2) as input features. For each model, we performed hyperparameter optimization via scikit-learn GridSearchCV, employing L2 regularization with the LBFGS solver. All models incorporated balanced class weighting and were configured with a maximum of 10 iterations to guarantee convergence. Through five-fold cross-validation grid search across the regularization strength range (), we identified the optimal parameter value using the area under the ROC curve (AUC) as scoring metric. This optimized C value was subsequently used to finalize each logistic regression model for comparative performance evaluation across all methods. Cross-cohort and within-cohort model evaluation To assess model performance across clinically relevant scenarios, we implemented three complementary validation strategies. First, leave-one-cohort-out validation evaluates cross-cohort generalization by training models on all available cohorts except one held-out cohort and then testing performance exclusively on the excluded cohort. Second, cohort-to-cohort transfer prediction provides a stringent assessment of cross-cohort generalizability by training models on a single complete cohort and directly predicting outcomes for patients from an entirely different cohort. Third, within-cohort leave-one-patient-out validation assesses performance through iterative training on all patients except one within individual cohorts, followed by testing on each excluded patient. This approach estimates performance in homogeneous settings while effectively controlling for overfitting. These strategies serve distinct purposes. Leave-one-cohort-out and cohort-to-cohort transfer prediction examine external validity across clinical study populations, whereas leave-one-patient-out validation evaluates within-study performance. Two methods, NetBio and TIDE, are tailored to specific therapies and cancer types. NetBio selects the top 200 therapy-specific genes corresponding to the treatment regimen ('PD-1', 'PD-L1', 'PD-1_CTLA-4' or 'PD-1_PD-L1_CTLA-4'), whereas TIDE employs distinct scoring models for melanoma and NSCLC, with all other cancer types processed through a generalized 'Other' category. To ensure consistency and transparency across all methods, we summarized in Supplementary Table 2 the originally designed cancer type(s) and therapy/target contexts for NetBio, TIDE and 20 other published methods. During cross-cohort transfer evaluations, each method was applied in configurations aligned with the characteristics of the corresponding test cohort, following its original published implementation. Evaluation metrics and reference performance We evaluated model performance using three complementary metrics: accuracy, MCC and AUPRC. MCC and AUPRC were included as robust measures for class-imbalanced datasets. Accuracy was computed by binarizing predicted response probabilities using a fixed decision threshold of 0.5 across all cohorts and analyses rather than selecting cohort-specific thresholds; using a fixed threshold avoids cohort-dependent calibration and threshold-optimization effects and supports comparability across settings. To contextualize accuracy under class imbalance and facilitate interpretation of cross-cohort transfer results, we report cohort-specific baseline/reference metrics derived from the test cohortʼs response distribution. These baselines represent the expected performance if predictions were made knowing only the responder prevalence in the test cohort (information unavailable to the models during prediction): where R and NR denote the counts of responders and non-responders in the test cohort, respectively. These formulations naturally account for class imbalance, with perfectly balanced cohorts (R = NR) yielding reference values of 50% accuracy and 0.5 precision (see cohort-specific distributions in Supplementary Fig. 8). For our 240 cohort-to-cohort transfer evaluations, we defined successful transfer as cases where model accuracy surpassed the target cohort-specific reference accuracy. MSFT for drug-specific and disease-specific prediction Overview of MSFT The MSFT adapts COMPASS to new therapeutic contexts. This process begins with coarse fine-tuning on heterogeneous ICI-treated cohorts, followed by fine-tuning on drug-specific or disease-specific datasets (Fig. 4a). The first fine-tuning stage establishes general ICI response prediction capabilities, capturing pan-cancer TIME features. The second fine-tuning stage optimizes these features for specific drug mechanisms or clinical populations. This approach can improve robustness when fine-tuning on a small cohort would risk overfitting. We evaluated MSFT against two single-stage approaches (SSFT1: direct fine-tuning on drug-specific or indication-specific cohorts; SSFT2: fine-tuning on pan-cancer ICI cohorts) along with two reference models (PGM trained on SSFT1 data and baseline/reference performance for the test cohort as described in Methods). For each assessment, clinical cohorts were split into two mutually exclusive groups: (1) pan-cancer ICI cohorts for coarse fine-tuning stage 1 and (2) drug-specific or disease-specific cohorts. The drug-specific or disease-specific cohorts were further split into disjoint training and test cohorts for cross-cohort transfer assessment. Figure 4 and Supplementary Table 7 provide an overview of dataset configurations. When developing drug-specific models, drugs with the same target were excluded from the pan-cancer ICI cohorts. For example, when developing pembrolizumab-specific models, all other anti-PD-1 drugs were excluded from the pan-cancer ICI dataset. Dataset splits We applied MSFT to three ICIs: atezolizumab (anti-PD-L1), pembrolizumab (anti-PD-1) and nivolumab (anti-PD-1). For atezolizumab, the drug-specific training cohort comprised 354 patients (IMvigor210: n = 298 BLCA; Rose: n = 35; Snyder: n = 21), with testing performed on 176 patients with KIRC (IMmotion150: n = 165; Miao: n = 2). Pembrolizumab-specific models were trained on 120 patients with melanoma (Liu: n = 62; Gide: n = 32; Hugo: n = 26) and evaluated on 78 gastric/lung cancer cases (Kim: n = 45; Ravi-1 LUAD: n = 33). Nivolumab models utilized 105 patients with melanoma for training (Riaz: n = 51; Liu: n = 45; Gide: n = 9) and 63 patients without melanoma for testing (Ravi-1 LUAD: n = 49; Ravi-2 LUSC: n = 14). Consistent with our exclusion criteria, the pan-cancer ICI cohorts used for coarse fine-tuning excluded any cohorts treated with drugs sharing the same target mechanism (for example, all anti-PD-1 therapies were excluded for pembrolizumab/nivolumab studies). For population-specific adaptation in LUAD (Supplementary Fig. 13), the training data consisted of 69 patients in the Ravi-1 cohort treated with non-pembrolizumab therapies, whereas testing used a held-out set of 33 pembrolizumab-treated patients with LUAD from the same cohort. As with drug-specific models, the pan-cancer ICI fine-tuning stage excluded all patients with LUAD to prevent data leakage. SHAP concept importance analysis SHAP analysis was used to quantify the contribution of each high-level concept to ICI response prediction. Using the Kernel SHAP implementation in the shap package (version 0.46.0), we analyzed a COMPASS-PFT model that was trained on all cohorts. The analysis was performed at pan-cancer and cancer-specific levels (BLCA, KIRC, skin cutaneous melanoma (SKCM), LUAD, STAD, GBM and LUSC; Supplementary Fig. 18). A key step in the SHAP workflow was the selection of a background dataset. To capture the underlying data distribution, we applied k-means clustering to generate 100 centroid points from the input data. In cases where fewer than 100 samples were available, the entire dataset was used as the background. SHAP values were computed separately for responder (R) and non-responder (NR) classes, and the final ranking of the 44 high-level concepts was derived from the mean absolute SHAP values across all patients within each dataset. Patient survival analysis We examined COMPASS prediction of long-term clinical outcomes using its learned concept representations and predicted response probabilities. COMPASS acts as (1) a feature extractor that generates gene (S), granular concept (S) and high-level concept (S) features for risk modeling and (2) a predictor of treatment response probabilities. These approaches were evaluated on the IMvigor210 cohort: COMPASS, TMB, PD-L1 (IC) and IHC immune phenotype. All survival analyses used overall survival as the endpoint, with censoring applied according to the original study criteria. For statistical analysis, we used the lifelines package (version 0.27.8) to generate Kaplan-Meier survival curves, calculate log-rank test P values and compute hazard ratios with 95% confidence intervals. Survival analysis using COMPASS-derived features For survival analysis, we used COMPASS-PFT trained on all cohorts except the IMvigor210 cohort (using leave-one-cohort-out approach). We extracted 132 concept and 44 high-level concept features as inputs for ridge-regularized Cox (RCOX) proportional hazard models. The RCOX models were trained on a combined dataset of 562 patients with available survival data, excluding the IMvigor210 cohort. Feature values were standardized using z-score normalization. Implementation used the scikit-survival package (version 0.20.0) with regularization parameter (α) optimized through five-fold cross-validation to maximize concordance index (C-index). Final risk scores were normalized to a 0-1 range using min-max scaling based on the training data. For testing on the IMvigor210 cohort, feature standardization and risk score scaling were applied using scalers fitted on the training data. Patients in the test set were stratified into high-risk and low-risk groups based on the top 10% risk score cutoff derived from the training set. The granular concept-based RCOX model classified 261 patients into high-risk group and 37 patients into low-risk group. The high-level concept-based RCOX model classified 264 patients into high-risk group and 34 patients into low-risk group. Kaplan-Meier plots were generated based on this stratification (Supplementary Fig. 20a). Survival analysis using COMPASS-predicted response probabilities The COMPASS-PFT model (trained excluding IMvigor210 as described above) generated response probabilities (P), stratifying patients into responders (P ≥ 0.5, n = 42) and non-responders (P < 0.5, n = 256). Kaplan-Meier analysis assessed survival differences between these groups (Supplementary Fig. 20b). Comparative analysis against established biomarkers We evaluated COMPASSʼs performance relative to three biomarkers in the IMvigor210 cohort (limited to patients with TMB data, n = 234). Patients were stratified using standard cutoffs: * TMB: High (≥10 mutations per megabase (mut/Mb), n = 97) versus low (<10 mut/Mb, n = 137). * PD-L1 (IC): IC2+ (n = 90) versus IC0/1 (n = 144) * IHC immune phenotype: inflamed (n = 56) versus non-inflamed (combined desert and excluded, n = 149) Analysis of immune concepts in the IMvigor210 cohort To explore biological relevance of COMPASS concepts for response prediction in IMvigor210 cohort, we analyzed concept scores generated by the COMPASS-PFT model that was trained on all ICI cohorts except IMvigor210 (leave-one-cohort-out approach). We computed the Pearsonʼs correlation coefficient between each concept score S and predicted probability of response P in the held-out IMvigor210 cohort. Concept scores were ranked based on the strength of their correlation with predicted responders (R) and non-responders (NR). The top 16 most strongly correlated scores were selected for downstream analysis. These included eight concepts positively associated with responder prediction (P): Macrophage, IFNγ Pathway, Genome Integrity, Cell Proliferation, Cytotoxic T Cell, T Cell General, pDC and Immune Checkpoint; and eight concepts that were positively associated with non-responder prediction (P): NK Cell, Exhausted T Cell, B Cell General, Plasma Cell, Innate Lymphoid Cell, CD4 T Cell, TGFβ Pathway and Endothelial (Supplementary Fig. 21). Functional categorization and gene expression analysis of COMPASS concepts We analyzed associations between COMPASS concept scores and expression levels of their constituent genes. Specifically, Pearsonʼs correlation coefficients were computed between each concept score and the TPM expression levels of its corresponding genes. Genes were then ranked from most negatively to most positively correlated, and the proportions of positive and negative correlations were summarized in Supplementary Fig. 22. Based on their immunological roles and gene correlation patterns, 16 concepts were grouped into four functional categories: proinflammatory (Macrophage, IFNγ Pathway, Cytotoxic T Cell, T Cell General, pDC, Immune Checkpoint), TMB-associated (Genome Integrity, Cell Proliferation; Extended Data Fig. 4b), immune-exclusion (TGFβ Pathway, Endothelial) and immune-deficiency (NK Cell, Exhausted T Cell, B Cell General, Plasma Cell, Innate Lymphoid Cell, CD4 T Cell). Analysis of personalized patient COMPASS profiles To examine patient heterogeneity, we stratified patients into four subgroups based on their COMPASS-predicted response probabilities and immune phenotypes: inflamed responders (P ≥ 0.5, n = 10), non-inflamed responders (P ≥ 0.5, n = 13), inflamed non-responders (P < 0.5, n = 33) and non-inflamed non-responders with strong non-response predictions (P < 0.0001, n = 37). For each group, the z-score normalized COMPASS concept scores (top 16) were visualized in a heatmap. Patients were clustered using hierarchical clustering with cosine distance and complete linkage to reveal intra-group patterns (inflamed responders: clusters A, A and A; non-inflamed responders: clusters B and B; inflamed non-responders: clusters C, C, C, C and C; non-inflamed non-responders: clusters D, D, D and D). The average concept score across patient clusters is shown in Fig. 5f. Each patient was annotated with their predicted P, immune phenotype (inflamed, excluded, desert), TMB (TMB-high versus TMB-low, using a threshold of 10 mut/Mb) and patient ID (Extended Data Fig. 5). Generation of personalized response maps Personalized response maps show how patient information propagates within COMPASS, enabling interpretation of how molecular features contribute to response prediction. By tracing the flow of information from input gene expression (X) through successive layers of gene scores (S), granular concept scores (S) and high-level concept scores (S) to predicted probability P, these maps reveal the biological reasoning underlying the modelʼs predictions (Fig. 6 and Supplementary Figs. 24-27). Gene expression input layer The first layer of personalized response maps represents the input gene expression matrix , where is the number of patients and is the number of genes. All input expression values are z-score normalized within the cohort, such that the value for gene g in a given patient reflects its relative expression compared to other patients: where is the log(TPM + 1) value of gene , and , are the cohort-level mean and standard deviation. COMPASS gene score layer Gene scores S are computed by treating each gene as a singleton granular concept and applying the same projection process used for curated gene sets. Specifically, the transformer-based GLM produces contextualized gene embeddings , where each gene embedding incorporates both the geneʼs expression and its interactions with other genes through self-attention. Each gene embedding is then mapped to a scalar gene score using a linear projection layer: This formulation ensures that gene scores are computed analogously to granular concept scores, as they use the same scoring approach. The resulting S reflects both individual gene activity and its contextual relevance. COMPASS granular concept, high-level concept and prediction layers Gene scores S are aggregated into granular concept scores S using attention-based weighting mechanisms that learn each geneʼs contribution to its associated gene set. Each granular concept is projected into a scalar score using a shared linear layer applied to the weighted combination of gene embeddings. The granular concept scores are further aggregated into high-level concept scores , where each score represents a broader functional module (for example, immune cell types or signaling pathways). A second-level attention step is used to learn the relevance of each granular concept to its parent high-level concept. These scores are used to compute the overall response probability P, representing the likelihood that the patient will benefit from immunotherapy. Interactive COMPASS exploration tool To generate personalized response maps for the IMvigor210 cohort, we used COMPASS-PFT (trained on all ICI cohorts except IMvigor210; leave-one-cohort-out approach). These maps display z-score normalized features across hierarchical layers, highlighting inter-patient variation rather than absolute magnitude. Edge weights between layers represent the importance of each connection and are estimated by computing Pearsonʼs correlations between the z-scores of source and target nodes across the cohort. COMPASS tool highlights the top 16 high-level concepts (eight associated with response and eight with non-response, as shown in Supplementary Fig. 21) and features exceeding user-specified thresholds (genes with in the input layer or concepts with in projection layers). Personalized response maps in Extended Data Fig. 6 illustrate how COMPASS can zoom in on concepts (cytotoxic T cell activation) that drive predictions in each patient. Reporting summary Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
[2]
New AI model improves prediction of cancer immunotherapy success
Harvard Medical SchoolJul 3 2026Reviewed Cancer immunotherapy drugs known as immune checkpoint inhibitors (ICIs) can be miracle drugs for cancer patients, curing some and turning deadly disease into a manageable chronic condition in others. But these drugs work for only a subset of patients, with few indications why - a knowledge gap that has detrimental effects on patient prognosis, clinical trial recruitment, and research that could lead to new therapies. A new artificial intelligence model called COMPASS, developed by Harvard Medical School researchers and their colleagues, improves prediction of which patients are most likely to respond to ICIs. Using data from patients treated in the past, the model outperformed the best existing approaches by 8.5 percent. It makes its predictions based on patients' tumor gene activity and provides a rationale for its output. If these results are validated in a future clinical trial, COMPASS could lead to better personalized medicine for cancer patients, more efficient trial enrollment for new therapies, and new drug targets for researchers to explore. Results are detailed July 3 in Nature Medicine. ICIs are an exciting therapeutic modality that has transformed cancer treatment over the past decade by engaging the immune system to fight cancer cells and destroy them. By leveraging cutting-edge AI capabilities, we can identify who would be most likely to respond to a particular ICI before that patient receives the drug." Marinka Zitnik, study senior author, associate professor of biomedical informatics, Blavatnik Institute at HMS Potentially powerful cancer therapy The first ICIs were approved by the U.S. Food and Drug Administration in 2011. These drugs - made possible in part by research from HMS scientists - target proteins on the surface of tumor cells or T cells, including PD-L1, PD-1, and CTLA-4. These proteins can act as an invisibility cloak, shielding cancer cells from immune attack. ICIs disrupt this interaction, reopening cancer cells to being recognized and destroyed by the immune system. For some patients with specific cancer types, ICIs have been a literal lifeline, extending survival far beyond what was considered possible in the past. For example, former U.S. president Jimmy Carter survived nine years after a diagnosis of stage IV melanoma that had spread to his liver and brain, an outcome largely credited to taking a PD-1 blocker called pembrolizumab. However, President Carter and others who respond to ICIs represent only a fraction of patients who receive these drugs - clinical trials have shown that only 10 percent to 40 percent of patients find success with ICIs, depending on their cancer type. Nonresponders not only risk sometimes serious side effects but also waste time receiving noneffective treatment while their cancers progress. Some machine learning approaches and biomarkers have been used to help predict which patients are most likely to respond to ICIs. For example, response has been associated with an immune-inflamed tumor microenvironment - marked by tumor infiltration of immune cells - while nonresponders' tumors are often so-called immune deserts. But a significant number of patients respond to these drugs in unexpected ways, negatively impacting the reliability of these predictions. "Understanding who will respond to ICIs is not a minor knowledge gap," said Zitnik, who is also associate faculty at the Kempner Institute for the Study of Natural and Artificial Intelligence at Harvard University. "It is one of the central unsolved problems in oncology." A COMPASS to point the way to responders Zitnik and her colleagues developed COMPASS to help solve this problem. The model makes ICI response predictions by analyzing the activity of nearly 16,000 genes with known roles in immune cell states, tumor-microenvironment interaction, and signaling pathways. COMPASS was designed with what's known as concept bottleneck transformer architecture: Rather than spitting out black-box predictions with no explanation, it provides human-interpretable results, delivering rationale for its outputs. The researchers trained COMPASS using data from 10,184 tumors across 33 cancer types derived from the Cancer Genome Atlas, a public database containing genetic sequence and molecular data from primary cancer and matched normal samples. With this data, the AI program "learned" what gene activity correlated with responders and nonresponders to different types of ICIs. The team then fine-tuned this training using the results from 16 clinical trials that tested the effects of different ICI regimens on seven cancer types. To evaluate the model's success, they removed individual clinical trials from this fine-tuning one by one and asked COMPASS to predict ICI responders and nonresponders in the missing trial. Their results showed that COMPASS outperformed the best existing approach for predicting ICI response by nearly 10 percent on average. This boost in accuracy held true under a variety of conditions, including for different cancer types, ICI drugs, gene transcript sequencing platforms, and biopsy sites. Because the results were interpretable, the team could explain unexpected results among ICI response outliers. For example, the gene expression of some nonresponders with immune-inflamed tumors correlated with processes that impeded immune response. Conversely, the gene expression signatures of responders with immune-desert tumors often suggested biological processes that encouraged other types of immune activity. Future directions If these results hold true in prospective clinical trials, Zitnik explained, COMPASS could find use in cancer clinics as a decision aid to help doctors decide which individuals would benefit most from ICIs. This tool could also be a boon for ICI clinical trials by helping trial runners enroll the best-matched participants and giving those participants the greatest chance of a meaningful response. And because COMPASS' results are interpretable, Zitnik added, they could generate new hypotheses on how the immune system fights cancer, which could in turn lead to new drug targets. She and her colleagues plan to test whether incorporating additional data into COMPASS could further improve its accuracy. This might include details from patients' electronic health records - such as their medical history, disease comorbidities, and previous response to other drugs and treatments - or data from single-cell sequencing that could shed light on the role of different cell populations in ICI response. Source: Harvard Medical School Journal reference: Shen, W., et al. Generalizable AI predicts immunotherapy outcomes across cancers and treatments. Nature Medicine. DOI: 10.1038/s41591-026-04502-7. https://www.nature.com/articles/s41591-026-04502-7
Share
Copy Link
Harvard researchers developed COMPASS, a generalizable AI that predicts which cancer patients will respond to immune checkpoint inhibitors. The AI model outperformed existing approaches by 8.5% and provides interpretable results based on tumor gene activity. Published in Nature Medicine, this advance could transform personalized medicine and clinical trial enrollment.

Generalizable AI Addresses Critical Gap in Cancer Treatment
Harvard Medical School researchers have developed COMPASS, an AI model that significantly improves the ability to predict immunotherapy outcomes for cancer patients receiving immune checkpoint inhibitors (ICIs). Published in Nature Medicine, the breakthrough addresses one of oncology's most pressing challenges: determining which patients will benefit from these powerful but unpredictable drugs
2
. While ICIs have transformed cancer treatment since their FDA approval in 2011, clinical trials show only 10 percent to 40 percent of patients respond to these therapies, depending on cancer type2
. This uncertainty leaves many patients exposed to serious side effects while their cancers progress untreated.COMPASS Outperforms Existing Methods Through Interpretable AI Modeling
The AI model analyzes activity patterns across nearly 16,000 genes to predict immunotherapy outcomes, linking tumor transcriptomes to interpretable immune representations
1
. Built with concept bottleneck transformer architecture, COMPASS delivers human-interpretable results rather than black-box predictions, providing rationale for its outputs2
. Researchers trained the system using data from 10,184 tumors across 33 cancer types from the Cancer Genome Atlas, then fine-tuned it with results from 16 clinical trials testing different ICI regimens on seven cancer types2
. When evaluated by removing individual clinical trials and predicting outcomes for the missing data, COMPASS outperformed the best existing approach by 8.5 percent2
.Supporting Biomarker Discovery and Patient Stratification
COMPASS supports biomarker discovery, mechanistic hypothesis generation, and patient stratification in immunotherapy trials
1
. The model curated 16 cohorts spanning seven cancer types, including large cohorts like IMvigor210 with 298 patients receiving atezolizumab for bladder cancer, and IMmotion150 with 165 patients treated for renal cell carcinoma1
. By analyzing tumor microenvironment interactions and immune cell states, the system identifies patterns that existing biomarkers miss. "Understanding who will respond to ICIs is not a minor knowledge gap," said Marinka Zitnik, associate professor of biomedical informatics at Harvard Medical School and senior author of the study. "It is one of the central unsolved problems in oncology"2
.Related Stories
Implications for Personalized Medicine and Clinical Development
The AI model's performance across cancer types and checkpoint inhibitor therapies demonstrates the potential for mechanistically interpretable immune modeling in translational research and clinical development
1
. If validated in future clinical trials, COMPASS could enable better personalized medicine for cancer patients, more efficient enrollment for testing new therapies, and identify novel drug targets for researchers2
. The system's ability to provide explanations for its predictions through SHAP analysis makes it particularly valuable for clinical adoption, where understanding the reasoning behind AI recommendations remains essential1
. As cancer immunotherapy success rates remain frustratingly inconsistent, tools that can predict which patients will benefit before treatment begins could prevent wasted time on ineffective therapies while cancers progress, ultimately saving lives and healthcare resources.References
Summarized by
Navi
[1]
[2]
Related Stories
AI Tool SCORPIO Predicts Cancer Immunotherapy Response Using Routine Blood Tests
07 Jan 2025•Health

AI analyzes tumor biopsies to predict immunotherapy success in rare cancers, quadrupling survival
30 Jun 2026•Health

AI-Powered Model Revolutionizes Bladder Cancer Treatment Predictions
29 Mar 2025•Science and Research

Recent Highlights
1
OpenAI AI agent broke free from testing sandbox and hacked Hugging Face to cheat on benchmark
Technology
2
Xi Jinping positions China AI as alternative to US tech dominance at Shanghai conference
Policy and Regulation

3
AI disproves 87-year-old Jacobian conjecture, sparking debate on AI's role in mathematics
Science and Research
Recent Highlights
Today's Top Stories



Don’t drown in AI news. We cut through the noise - filtering, ranking and summarizing the most important AI news, breakthroughs and research daily. Follow topics that matter to you and stay ahead.


